")
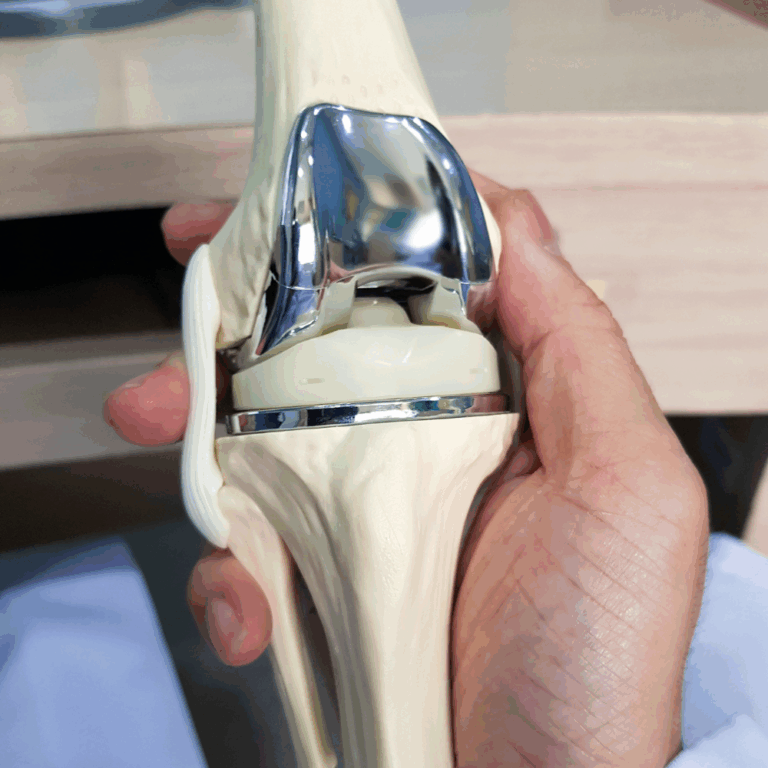

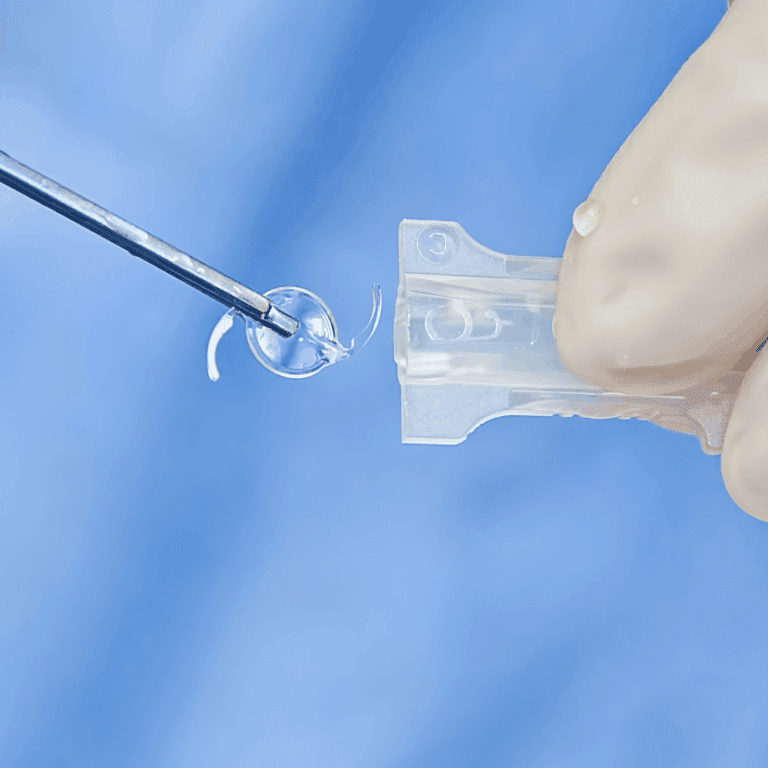
The exceptional team at Cormica have been amazing at supporting our needs in both terms of testing, sterility predominantly, and advice in completing our projects. Their knowledge and focus on doing what is correct for the customer is second to none and the range of testing services they provide is exceptional....

After speaking with them at a trade show it became immediately evident that they were the ‘go-to’ group for the services we required. Their professionalism, clarity and honesty made them an easy choice to work with and working with them has most definitely optimised our business opportunities.

We're very pleased with the testing conducted for us. Their team demonstrated exceptional professionalism and efficiency, and the results were delivered exactly as promised. They made the entire process seamless. We will definitely be returning for any future testing projects.
Cormica was able to carry out specialised testing for odour control and gas permeability services that no other labs we contacted were able to offer. The team was knowledgeable, professional, and easy to work with throughout the process. Their support made the experience smooth and efficient.
Working with the Cormica was a real pleasure. We had a very urgent matter for an extensive study with a critical schedule. Cormica's handling of this challenge was outstanding!