
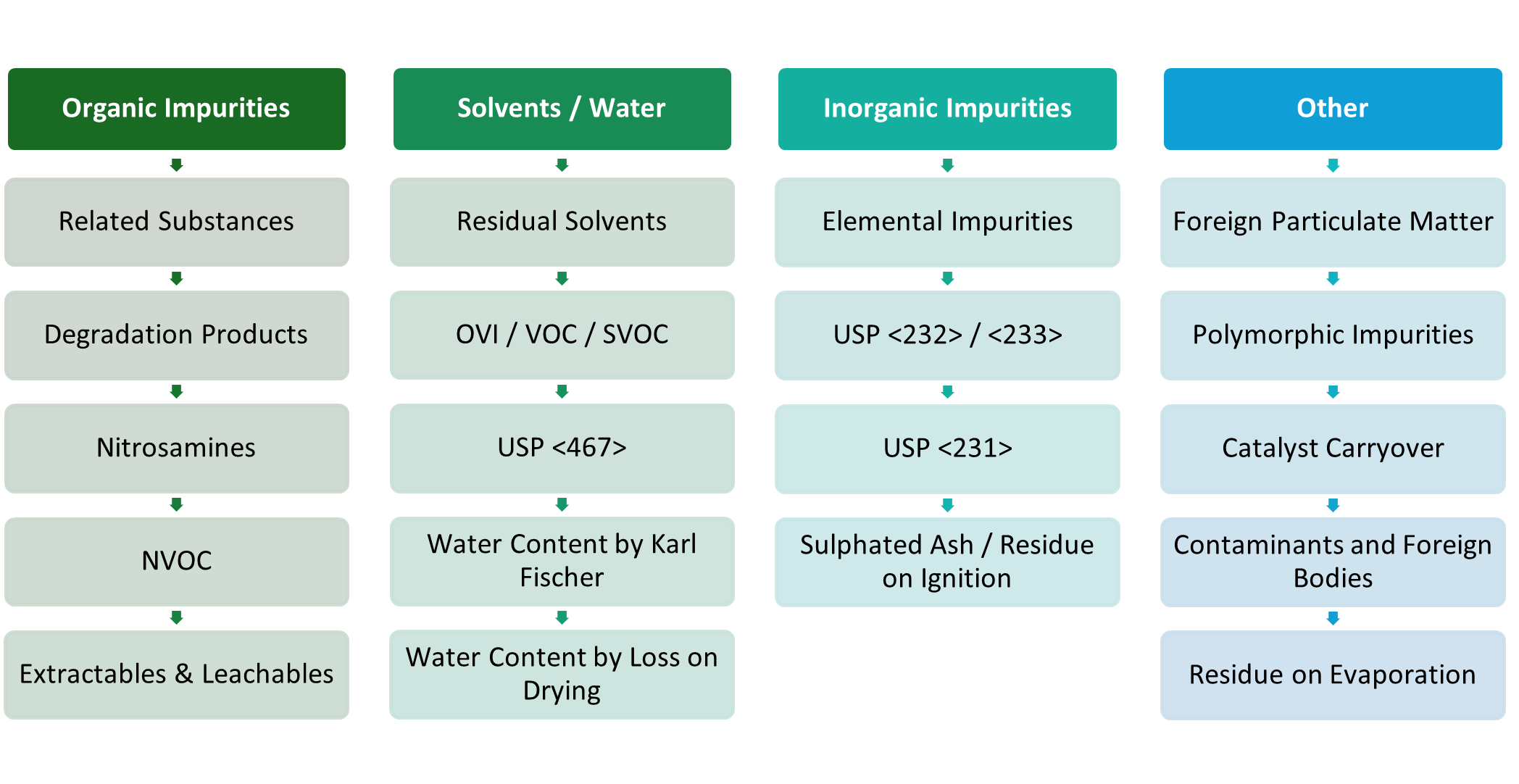
Identification and control of organic impurities in a drug substance and drug product is critical to the quality and safety of any drug product. Out of specification results and contamination can have huge cost and time implications for new products reaching the market or the batch release of existing products.
Organic impurities can arise in many ways, whether from residual excipients or starting materials, degradation products or contamination, and the reporting and control of these impurities is a fundamental expectation of regulatory authorities, including the guidelines ICH Q3A/B, European Pharmacopoeia (Ph. Eur.) chapter <5.10> and US Pharmacopeia (USP) general chapter <1086>.
At Cormica we recognise that reliable detection of both organic impurities and contamination can be challenging and as such we have a range of different detectors coupled with our (U)HPLC instruments, which facilitate detection of many different and challenging molecules:

Nitrosamines are a specific type of organic impurity containing a nitroso functional group; they represent potent mutagenic carcinogens and have been classified as probable human carcinogens by World Health Organisation and other health authorities. Consequently, the Food and Drug Administration (FDA), European Medicines Agency (EMA) and other regulatory agencies require all relevant finished product and medical device manufacturing licence holders to risk assess the potential for nitrosamine contamination in their products. If risk assessments indicate potential for contamination, or confirmation of trace levels below specification limits is required, screening and testing products for the presence of nitrosamines has become vital.
Here at Cormica, we provide a comprehensive nitrosamine screening service, along with nitrosamines identification and quantitation, tailored to meet the needs of drug substance, finished product and medical device manufacturers. This is carried out via a strategic / tailored approach, based on your requirements, and using instrumentation designed to achieve the lowest detection levels.

With many years of experience, our scientists are adept at the development and validation of bespoke analytical procedures, while also delivering our in-house developed / validated analytical method.
We pride ourselves in the depth of knowledge in our team and our ability to overcome the challenges presented by low detection levels and challenging sample matrices. Through meticulous sample preparation techniques with an aim of isolation of test analytes from simple to challenging matrices coupled with LC-QQQ and GC-QQQ capabilities we are confident we can aid you in your product impurity assessments.
Packaging, storage and distribution of products, plus the interaction of the product with the packaging, along with the stability of the product during storage and distribution, are important parameters to investigate when seeking to understand the quality of your product and its potential to introduce unwanted substances to the human body.
Cormica offers a comprehensive suite of testing to support your assessment of biocompatibility, encompassing drug products, medical devices, advanced wound management and combination products.
Moreover, a key part of the biocompatibility risk assessment described in ISO 10993 involves assessing levels of both Extractable and Leachable compounds:
- Extractables profiling assesses and aims to identify any compounds released from a medical device, wound dressing or packaging item under exaggerated clinically relevant conditions (e.g. elevated temperature, aggressive solvents, extended time). This is considered the worst-case scenario, in terms of the generation of hazardous unknowns.
- Leachables assessment involves simulating the “real life scenario” use of any medical device, wound dressing or packaging item, to determine the potential for the patient to be exposed to the hazardous unknowns identified during the extractables phase, under normal storage conditions or during the product’s intended use.
The hazardous unknowns could include volatile, non-volatile or semi-volatile organic compounds, elemental and inorganic materials, and others, based on their relevance to the medical device, packaging, materials of construction and manufacturing process.
The identification and quantitation of such species supports an informed toxicological risk assessment and biological evaluation, including Analytical Evaluation Threshold (AET). ICH guidelines and ISO 10993, along with USP general chapters <1663> and <1664>, which are specific to Pharmaceutical Packaging / Delivery Systems, are key guidelines for such studies.
Organic solvents are often used in the manufacture or purification of drug substances, excipients or drug products but due to their potential toxicity their absence/presence must be verified in the pharmaceutical products to ensure patient safety. These solvents are evaluated for their possible risk to human health and are placed into one of three classes based on their toxicity data and their environmental impact as shown.
The classification determines the permissible limit of the solvent, based on permitted daily exposure (PDE) and a maximum 10 g daily dose.
USP general chapter <467> Residual Solvents provides detailed procedures for screening, confirmation and quantitation of residual solvents, including sample preparation and analytical conditions, and is harmonised with limits set out in the ICH Q3C Guideline for Residual Solvents (and also Ph. Eur. <5.4>, Residual Solvents)
At Cormica, we have extensive experience of establishing USP <467> compliant residual solvents methods for our customers; alternatively, we can transfer in your validated method. Additionally, our scientists can validate / verify our in-house Headspace GC-FID methodology for your product; using this approach, up to 52 different residual solvents may be identified and quantified in pharmaceutical actives, excipients, powder formulations and finished products, and others. For finished product, the client may choose to test either all the individual components or the final finished product.
Residual Solvents Classification and Risk Assessment (ICH Q3C)
| Residual Solvents Classification | Risk Assessment | Examples |
| Class 1 Solvents to be avoided | Known human carcinogens | Benzene Carbon Tetrachloride 1,1-Dichloroethane 1,1,1-Trichlorethane |
| Strongly suspected human carcinogens | ||
| Solvents with environmental hazard properties | ||
| Class 2 Solvents to be limited | Nongenotoxic animal carcinogens or agents of irreversible toxicity | Chloroform Cyclohexane Methanol Tetrahydrofuran |
| Solvents suspected of other significant but reversible toxicities | ||
| Class 3 Solvents with low toxic potential | Solvents with low toxic potential; 50 mg per day or less is acceptable | Acetone Ethanol Heptane Methyl Ethyl Ketone |
| Class 4 Solvents with insufficient data | Solvents without adequate toxicological data; justification is required | Isooctane Methylisopropyl ketone Petroleum Ether Trifluoroacetic acid |
Water content determination is a key factor in manufacturing processes and quality assurance, as the level of water present greatly influences the physical properties of a product at all stages of the manufacturing cycle.
Pharmaceuticals have the potential to contain bound and unbound (free) water, or to exist in hydrated forms, and these different manifestations of water content have significantly different impacts on stability, processability and drying. Hence, determining the water content (and type / location of water present) is essential to ensure compliance with regulatory standards.
At Cormica we offer a range of gravimetric and chemical techniques to suit your requirements.
- Loss on Drying: Performed according to US Pharmacopeia (USP) and European Pharmacopeia (Ph. Eur.) guidelines, utilising a drying oven with or without vacuum.
- Thermogravimetric Analysis: Our Perkin Elmer TGA 8000 offers an in-depth analysis of how a product or material’s mass changes when subjected to varying temperatures over different time intervals. The resulting thermograms reveal the material’s changes, pinpointing the exact temperatures at which these changes occur. The versatility of this technique allows for the testing of a broad variety of sample types.
- Karl Fischer (KF) Titration: The most commonly used method for determining water content due to its speed and high selectivity, it is applicable to a wide range of sample types, with water concentrations ranging from ppm to 100 % w/w levels. With our Metrohm 852 Combined Karl Fischer with 860 Oven, Cormica offers tailored testing services to meet specific needs of your samples using the following methods:
- Volumetric Karl Fischer: A direct titration technique suitable for solid and liquid samples containing 0.1-100 % w/w of water.
- Coulometric Karl Fischer: This method uses an electric current to determine the water content of liquid or dissolved solid samples, with a water content ranging from 0.001-1 % w/w.
- Karl Fischer Oven: If a sample cannot be dissolved in a suitable solvent or reacts with KF reagents, it may be heated in a sealed vial to volatilise the water into the headspace. This is then sampled and transferred to the coulometric vessel for water determination.
The ICH issued its Guideline for Elemental Impurities (ICH Q3D) in 2018. This was a step change away from classical wet chemistry methods, and towards element specific testing, and is also detailed in USP general chapters <232> and <233> and equivalent chapters in the European Pharmacopoeia (and others).
Permitted daily exposure (PDE) limits-based administration route and a maximum 10 g daily dose are provided in the guidance documents for 24 elements.
The guidance stresses the importance of risk assessing all potential sources of introduction of elemental impurities including:

With a diverse range of preparation equipment available onsite and both ICP-OES and ICP-MS instruments to perform the testing, our experienced staff will design your method to meet the specific challenges of your matrix, and the required limits set out in the guidance documents.
We collaborate to achieve an appropriate study design to meet your specific needs and efficiently deliver a compliant program.
Where necessary, Cormica can support with ongoing analysis of specific excipients, drug substances, drug product or container closure systems to demonstrate continuing process and product control.
We understand the need to continue to support our customers with legacy methodologies that may already be registered for their product, and we continue to offer a full range of wet chemistry methods, including heavy metals to USP general chapter <231>.
In addition to elemental impurities, a number of other inorganic impurities can be present and require identification and quantitation in pharmaceutical actives, excipients, powder formulations and finished products, amongst other materials. These can include residual reagents, ligands and inorganic salts.
Using our extensive range of instruments, we can develop and validate a bespoke and targeted methodology for your inorganic impurity; we continue to offer USP general chapter <281> Residue on Ignition (Sulphated Ash) and Ph. Eur. chapter <2.4.14>.
This non-specific test measures the amount of residual substance not volatilised from a sample when the sample is ignited in the presence of sulfuric acid and is used for determining the total content of inorganic impurities in an organic substance.
Polymorphic Impurities
Our scientists apply X-ray Powder Diffraction (XRPD) and Differential Scanning Calorimetry (DSC) to detect and quantify the polymorphic form(s) of key active and inactive ingredients throughout the development cycle. Different crystal forms of the same drug molecule can have varying properties such as melting point, solubility, dissolution rate, and bioavailability, which directly impact the safety, efficacy, stability, and the processability, of the API and finished product.
Understanding and controlling polymorphism, and monitoring polymorphic impurity content, is crucial for ensuring consistent drug performance, optimizing drug development, and maintaining patient safety.
At Cormica polymorphic purity / impurity determinations are used to monitor and control the quality of client’s drug substance and drug product via validated qualitative, limit or fully quantitative methods, as described in USP general chapter <941> Characterization of Crystalline and Partially Crystalline Solids by X-Ray Powder Diffraction.
Our experienced scientists can also use these techniques to support your process development and process control, and help you understand the drivers behind polymorphic change via polymorphic risk assessment or a polymorph screen.
Foreign particulate matter (FPM), present in parenteral injections, infusions, ophthalmic preparations and others, is defined as mobile, undissolved particles, other than gas bubbles, which are unintentionally present. The presence of foreign matter can arise through the manufacturing, preparation or packaging process and can have serious consequences for the quality and effectiveness of the product – and for patient safety.
One of the primary guidelines for injections is USP general chapter <788> Particulate Matter in Injections. This specifies the acceptance criteria for the number of particles present in the majority of small- and large-volume parenteral injections. In particular, the focus is on sub-visible particles, i.e., particles of ˂ 100 µm in size. Different classifications of particle size are presented, dependant on the injection volume.
At Cormica, we provide both light obscuration (Method I) and microscopic (Method II) particle counting, plus identification of foreign particles using the Malvern Panalytical Morphlogi4-ID. Our team uses the APPS 2000 light obscuration particle sizer, which is designed to meet pharmacopeial requirements and supports the testing of a wide range of liquids and suspensions. It can be used for determining the particle count in a sample over the range of particle sizes from 2-125 µm and is capable of measuring sample volumes from 0.2 mL to 1 litre.






